Alchemical Protein Mutations¶
Introduction¶
In this tutorial you will learn how to use BioSimSpace’s mapping functionality to set up alchemical calculations in order to compute the change in the binding affinity of a ligand as a result of a protein mutation. Specically, we are going to focus on two proteins, first a set up of a single alchemical point mutation on ubiquitin, and second a set up on aldose reductase (AR), which is a drug target for the treatment of diabetic nephropathy. It is recommended to complete previous BioSimSpace tutorials before attempting this one.
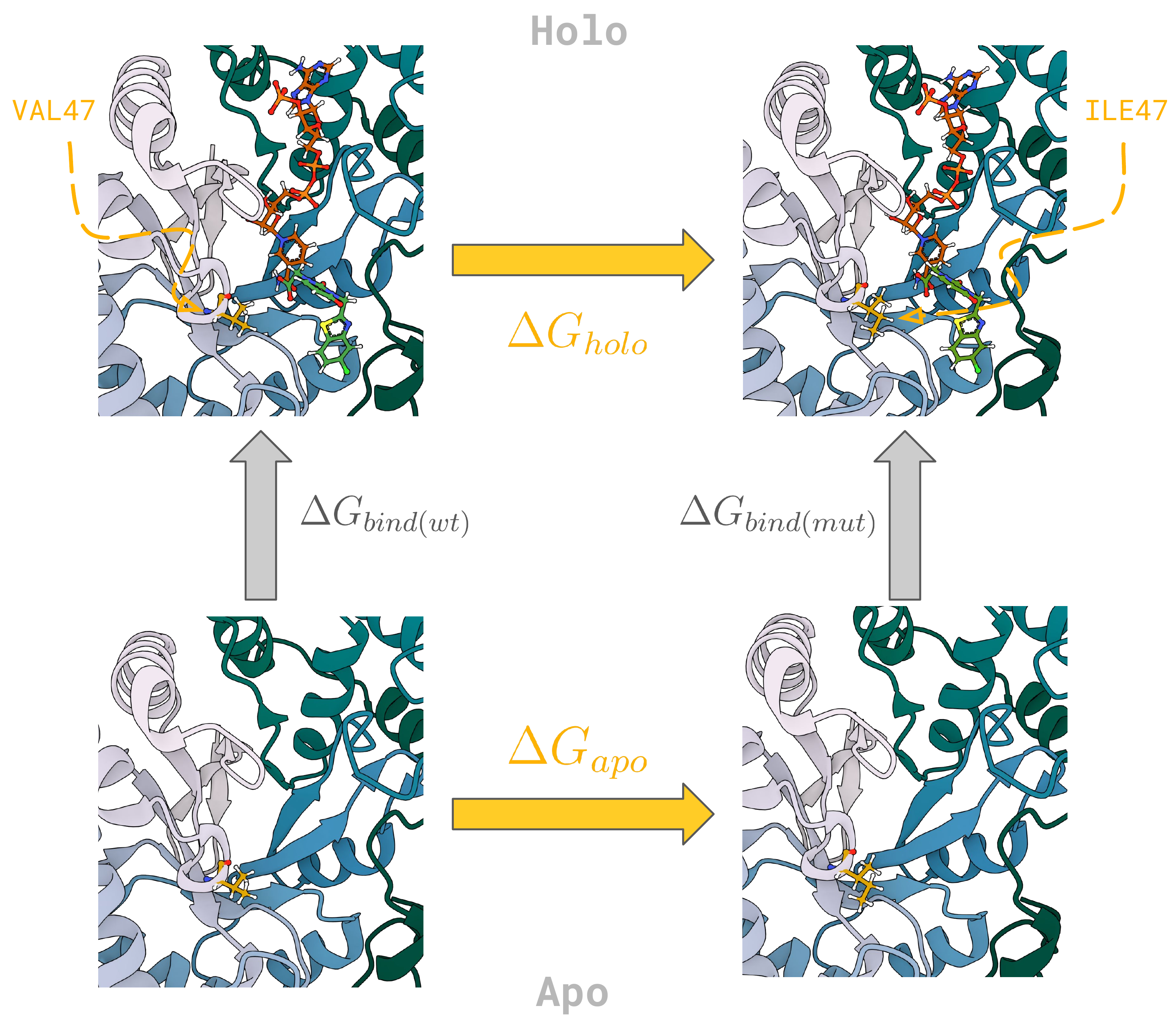
The relative change in the binding affinity as a result of a mutation, \(\Delta \Delta G_{mut}\) can be calculated from the difference between free energy of mutation in the holo (bound) and apo (unbound) simulation legs, i.e.:
To get started, let’s go through a simple example of generating the required input files in order to set up an alchemical mutation.
Simple Case - Input File Generation¶
In order to create an alchemical protein system in BioSimSpace, we need
two input protein structures, a wild-type and a mutant. We also need to
make sure that the atom ordering between the two proteins is identical.
Don’t worry, this is an easy assumption to satisfy. We will load a
structure 1UBQ via sire, which
comes with bundled with BioSimSpace:
import BioSimSpace as BSS
import sire as sr
mols = sr.load("1UBQ")
INFO:rdkit:Enabling RDKit 2024.03.3 jupyter extensions
INFO:numexpr.utils:Note: NumExpr detected 20 cores but "NUMEXPR_MAX_THREADS" not set, so enforcing safe limit of 8.
INFO:numexpr.utils:NumExpr defaulting to 8 threads.
Using cached download of 'https://files.rcsb.org/download/1UBQ.pdb.gz'...
Using cached unzipped file './1UBQ.pdb'...
There are multiple of ways of generating a mutant structure from a wild-type protein, some examples are: - Pymol Mutagenesis Plugin (when exporting the mutant structure, you want to make you select ‘retain atom ids’ under ‘PDB Options’, or pass both input structures through pdb4amber) - HTMD - FoldX - pdb4amber
For this simple case we are going to use pdb4amber to mutate a threonine at position 9 to an alanine residue. First we are going to pass the wild-type protein from the crystal structure through pdb4amber in order create a consistent atom ordering between wild-type and mutant structures:
!pdb4amber --reduce --dry --add-missing-atoms -o 1UBQ_dry_wt.pdb 1UBQ.pdb
==================================================
Summary of pdb4amber for: 1UBQ.pdb
===================================================
----------Chains
The following (original) chains have been found:
A
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
---------- Missing heavy atom(s)
None
Next, we are going to create a mutant structure:
!pdb4amber --reduce --dry -o 1UBQ_dry_t9a.pdb -m "9-ALA" --add-missing-atoms 1UBQ_dry_wt.pdb
==================================================
Summary of pdb4amber for: 1UBQ_dry_wt.pdb
===================================================
----------Chains
The following (original) chains have been found:
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
---------- Missing heavy atom(s)
ALA_9 misses 1 heavy atom(s)
Warning: This is a simple, but ultimately a crude way of generating a mutant structure. Different factors such as sidechain rotomers, packing and protonation states need to be taken into the account in order to accurately and robustly describe the mutant end-state.
Simple Case - Alchemical System Generation¶
Now that correct input files have been created, we can now proceed to create an alchemical protein in BioSimSpace. Let’s load our two proteins:
protein_wt = BSS.IO.readMolecules("1UBQ_dry_wt.pdb")[0]
protein_mut = BSS.IO.readMolecules("1UBQ_dry_t9a.pdb")[0]
Next, we want to parametrise them with our forcefield of choice:
protein_wt = BSS.Parameters.ff14SB(protein_wt).getMolecule()
protein_mut = BSS.Parameters.ff14SB(protein_mut).getMolecule()
Now we want to compute the mapping between the two proteins, first let’s figure out the residue index of our residue of interest (ROI):
protein_wt.getResidues()[7:10]
[<BioSimSpace.Residue: name='LEU', molecule=5, index=7, nAtoms=19>,
<BioSimSpace.Residue: name='THR', molecule=5, index=8, nAtoms=14>,
<BioSimSpace.Residue: name='GLY', molecule=5, index=9, nAtoms=7>]
protein_mut.getResidues()[7:10]
[<BioSimSpace.Residue: name='LEU', molecule=7, index=7, nAtoms=19>,
<BioSimSpace.Residue: name='ALA', molecule=7, index=8, nAtoms=10>,
<BioSimSpace.Residue: name='GLY', molecule=7, index=9, nAtoms=7>]
We can see that the residue with the index value of 8 are different between the two proteins. Let’s pass this value to the BioSimSpace.Align.matchAtoms function:
mapping = BSS.Align.matchAtoms(molecule0=protein_wt, molecule1=protein_mut, roi=[8])
Note: You can also pass multiple residues of interest indices to the mapping if you wish to mutate several residues simultaneously.
Now that the mapping has been computed, we can visualise it:
BSS.Align.viewMapping(protein_wt, protein_mut, mapping, roi=8, pixels=500)
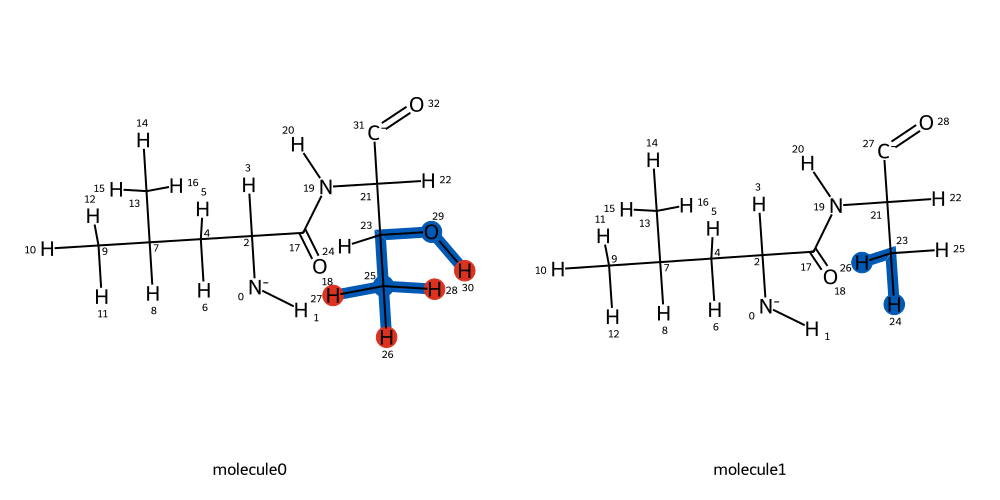
The computed atom mapping shows that both hydroxyl and methyl groups in the threonine side chain will be transformed into hydrogen atoms respectively. We can now proceed to align the two residues of interest:
aligned_wt = BSS.Align.rmsdAlign(molecule0=protein_wt, molecule1=protein_mut, roi=[8])
Finally, we can create a merged alchemical protein system:
merged_protein = BSS.Align.merge(aligned_wt, protein_mut, mapping, roi=[8])
The alchemical protein can now be solvated, ionised and exported to different file formats, for example GROMACS or SOMD2, our OpenMM-based FEP engine:
merged_system = merged_protein.toSystem()
# solvate the system with the padding of 15 angstroms
padding = 15 * BSS.Units.Length.angstrom
box_min, box_max = merged_system.getAxisAlignedBoundingBox()
box_size = [y - x for x, y in zip(box_min, box_max)]
box_sizes = [x + padding for x in box_size]
box, angles = BSS.Box.rhombicDodecahedronHexagon(max(box_sizes))
solvated_system = BSS.Solvent.tip3p(molecule=merged_system, box=box, angles=angles, ion_conc=0.15)
# export the solvated system to GROMACS input files
BSS.IO.saveMolecules("apo_ubiquitin_t9a", solvated_system, ["gro87", "grotop"])
# export the solvated system to SOMD2 input file
BSS.Stream.save(solvated_system, "apo_ubiquitin_t9a")
Aldose Reductase - Alchemical System Generation¶
Apo System¶
Now we are going to focus on the aldose reductase system and set up an alchemical transformation in both apo and holo forms of the protein. The input files (2PDG_8.0) were taken from the SI of a paper by Aldeghi et. al, residue 47 mutated via PyMol (V47I), and standardised via pdb4amber.
protein_wt = BSS.IO.readMolecules(BSS.IO.expand(BSS.tutorialUrl(), "aldose_reductase_dry.pdb"))[0]
protein_mut = BSS.IO.readMolecules(BSS.IO.expand(BSS.tutorialUrl(), "aldose_reductase_v47i_dry.pdb"))[0]
We can use ensure_compatible=False in order to get tLEaP to re-add
the hydrogens for us:
protein_wt = BSS.Parameters.ff14SB(protein_wt, ensure_compatible=False).getMolecule()
protein_mut = BSS.Parameters.ff14SB(protein_mut, ensure_compatible=False).getMolecule()
This time we are going to automatically detect the different residues between the two proteins:
roi = []
for i, res in enumerate(protein_wt.getResidues()):
if res.name() != protein_mut.getResidues()[i].name():
print(res, protein_mut.getResidues()[i])
roi.append(res.index())
<BioSimSpace.Residue: name='VAL', molecule=22664, index=45, nAtoms=16> <BioSimSpace.Residue: name='ILE', molecule=22666, index=45, nAtoms=19>
We can then pass these residue indices to the mapping function as before:
mapping = BSS.Align.matchAtoms(molecule0=protein_wt, molecule1=protein_mut, roi=roi)
BSS.Align.viewMapping(protein_wt, protein_mut, mapping, roi=roi[0], pixels=500)
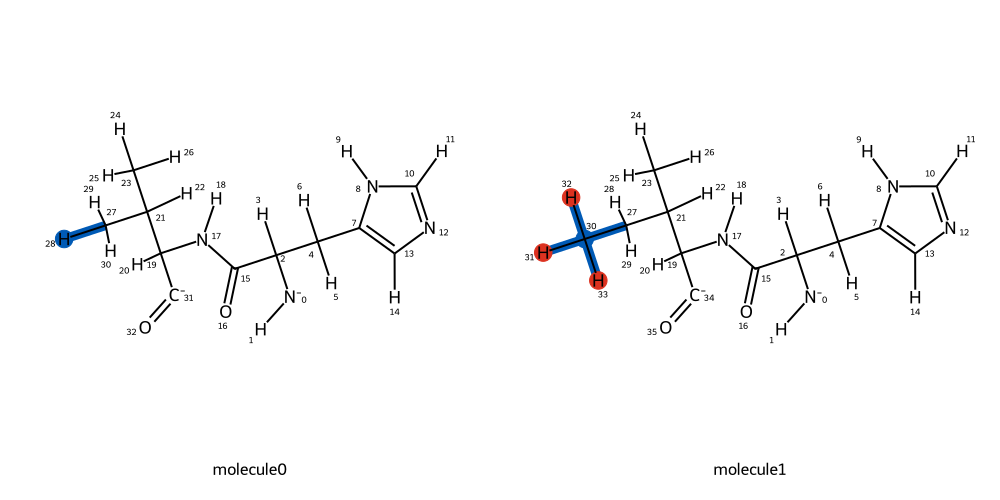
The mapping shows that the perturbation will transform a hydrogen to a methyl group. Is this what we would expect for a valine to isoleucine transformation? If we are happy, we can proceed with the rest of the set up as before:
aligned_wt = BSS.Align.rmsdAlign(molecule0=protein_wt, molecule1=protein_mut, roi=roi)
merged_protein = BSS.Align.merge(aligned_wt, protein_mut, mapping, roi=roi)
merged_system = merged_protein.toSystem()
padding = 15 * BSS.Units.Length.angstrom
box_min, box_max = merged_system.getAxisAlignedBoundingBox()
box_size = [y - x for x, y in zip(box_min, box_max)]
box_sizes = [x + padding for x in box_size]
box, angles = BSS.Box.rhombicDodecahedronHexagon(max(box_sizes))
solvated_system = BSS.Solvent.tip3p(molecule=merged_system, box=box, angles=angles, ion_conc=0.15)
BSS.IO.saveMolecules("apo_aldose_reductase_v47i", solvated_system, ["gro87", "grotop"])
Holo System¶
To set up a holo (bound) system, we are going to load in the associated ligand and the cofactor of aldose reductase:
ligand_47d = BSS.IO.readMolecules(BSS.IO.expand(BSS.tutorialUrl(), ["ligand_47_gaff2.gro", "ligand_47_gaff2.top"]))[0]
cofactor_nap = BSS.IO.readMolecules(BSS.IO.expand(BSS.tutorialUrl(), ["cofactor_nap_gaff2.gro", "cofactor_nap_gaff2.top"]))[0]
We can use BioSimSpace’s AMBER parametrisation pipeline if we wish to, but in this case the ligands have been parametrised for us so we can skip the following cell:
ligand_47d = BSS.Parameters.gaff2(ligand_47d, charge_method="BCC", net_charge=-1).getMolecule()
cofactor_nap = BSS.Parameters.gaff2(cofactor_nap, charge_method="BCC", net_charge=-4).getMolecule()
We can simply add the ligands to our alchemical protein in order to create an alchemical holo system. This way we are assuming that the ligands are already placed correctly with respect to the protein:
merged_system = merged_protein + ligand_47d + cofactor_nap
As before we can now proceed to solvate, ionise and export our prepared system or use BioSimSpace’s functionallity to further set up and execute the alchemical simulations.
padding = 15 * BSS.Units.Length.angstrom
box_min, box_max = merged_system.getAxisAlignedBoundingBox()
box_size = [y - x for x, y in zip(box_min, box_max)]
box_sizes = [x + padding for x in box_size]
box, angles = BSS.Box.rhombicDodecahedronHexagon(max(box_sizes))
solvated_system = BSS.Solvent.tip3p(molecule=merged_system, box=box, angles=angles, ion_conc=0.15)
BSS.IO.saveMolecules("holo_aldose_reductase_v47i", solvated_system, ["gro87", "grotop"])
Advanced Case - Bond Creation/Annihilation Transformations¶
In this tutorial we will use BioSimSpace’s mapping functionality to set up alchemical calculations involving proline mutations in a protein. Specifically, we will look at the Leu-to-Pro mutations in OMTKY3 to its receptors as detailed in this study.
wt = BSS.IO.readMolecules(
BSS.IO.expand(BSS.tutorialUrl(), f"1choFH_apo_wt_flare_processed.pdb")
)[0]
mut = BSS.IO.readMolecules(
BSS.IO.expand(BSS.tutorialUrl(), f"1choFH_apo_mut_flare_processed.pdb")
)[0]
wt = BSS.Parameters.ff14SB(wt, ensure_compatible=False).getMolecule()
mut = BSS.Parameters.ff14SB(mut, ensure_compatible=False).getMolecule()
Comparing the residues between two proteins shows us that the residues at index 15 are different between the proteins
roi = []
for i, res in enumerate(wt.getResidues()):
if res.name() != mut.getResidues()[i].name():
print(res, mut.getResidues()[i])
roi.append(res.index())
<BioSimSpace.Residue: name='LEU', molecule=5, index=15, nAtoms=19> <BioSimSpace.Residue: name='PRO', molecule=7, index=15, nAtoms=14>
By default, the
BioSimSpace.Align.matchAtoms
would fail to create a mapping for the ROI region, as the underlying
RDKit MCS algorithm would be unable to determine a mapping between two
molecular graphs with a fundamental topological mismatch. Because
Proline’s sidechain forms a cyclic system with the backbone, its atoms
exist in a ring topology. The algorithm restricts the mapping of cyclic
atoms to acyclic atoms (a behavior governed by parameters like
ringMatchesRingOnly) to preserve the chemical integrity of the
substructure. Consequently, a 1:1 mapping between the ring-bound
sidechain of Proline and the acyclic sidechain of the Leucine residue
cannot be determined.
Instead we can use the custom_roi_map argument of the
BioSimSpace.Align.matchAtoms
to override the RDKit MCS mapping. For example we can force an empty
mapping between the two residues:
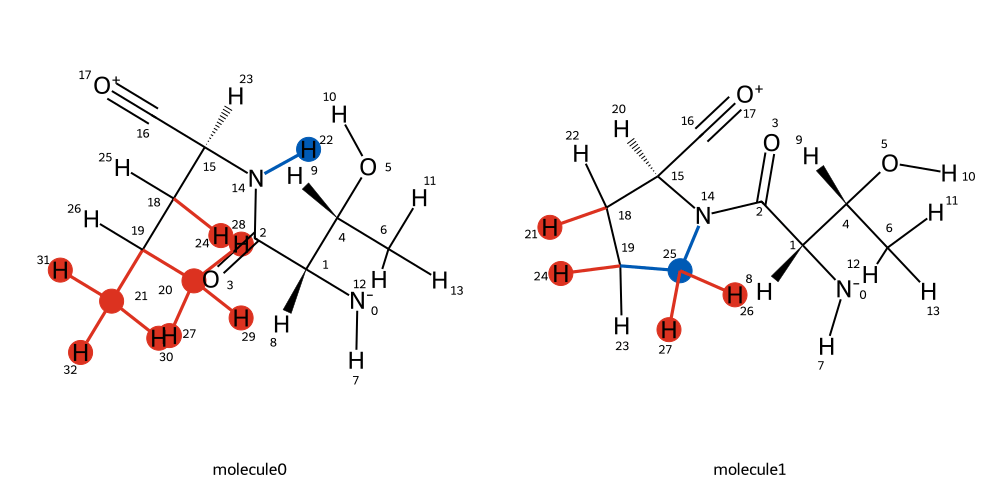
mapping = BSS.Align.matchAtoms(molecule0=wt, molecule1=mut, roi=[15], custom_roi_map={})
BSS.Align.viewMapping(wt, mut, mapping, roi=15, pixels=500)
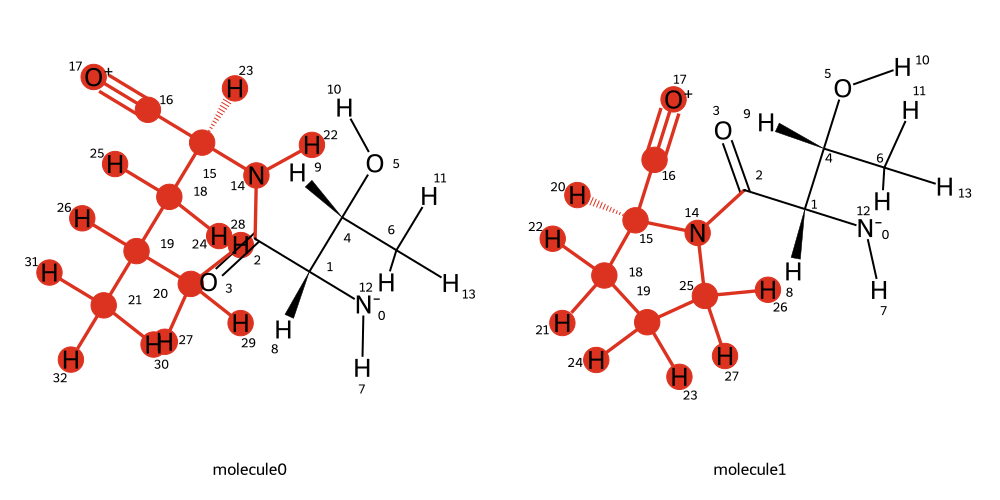
If we know the correct 1:1 atom mapping between the two residues, we can
pass that to the custom_roi_map which will allows us to setup an
alchemical bond transformation for mutating the leucine residue to
proline. Note that absolute atom indices need to be passed, i.e the
indices of the residues in the context of the whole protein. We can
use something like PyMol to help us map the atoms in the right order:
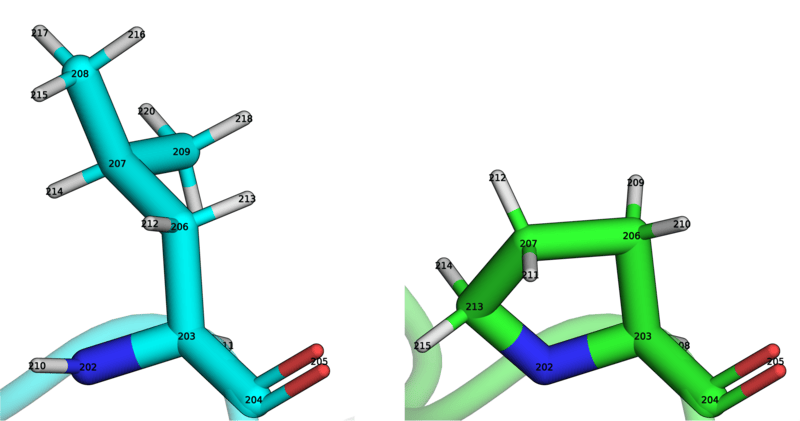
mapping = BSS.Align.matchAtoms(molecule0=wt, molecule1=mut, roi=[15], custom_roi_map={204:204,205:205,203:203,202:202,211:208,206:206,213:210,207:207,214:211,210:213})
BSS.Align.viewMapping(wt, mut, mapping, roi=15, pixels=500)
We can then use allow_ring_breaking=True argument of the
BioSimSpace.Align.merge
to create the required alchemical transformation:
aligned_wt = BSS.Align.rmsdAlign(molecule0=wt, mapping=mapping, molecule1=mut)
merged_protein = BSS.Align.merge(aligned_wt, mut, mapping, allow_ring_breaking=True, roi=[15])
But how do we actually know that the merge has built a perturbable molecule that now has a bond annihilation or creation involved? We can use Sire’s conversion features to check what kinds of alchemical modifications are happening in our perturbable molecule. You can check out the corresponding Sire tutorial for more details.
merged_protein_sire = merged_protein._sire_object
pert = merged_protein_sire.perturbation()
pert_omm = pert.to_openmm(map={"coordinates":"coordinates0"})
pert_omm.changed_bonds(to_pandas=True)
| bond | length0 | length1 | k0 | k1 | |
|---|---|---|---|---|---|
| 0 | N:203-H:211 | 0.1010 | 0.1449 | 363171.2 | 282001.6 |
| 1 | CG:208-H:211 | 0.1526 | 0.1526 | 0.0 | 259408.0 |
By comparing the k0 and k1 values in the changed bond dataframe,
we can see that the transformation is going to result in a bond being
created.